KinCoRe
Kinase Conformation Resource
News - February 19, 2026.
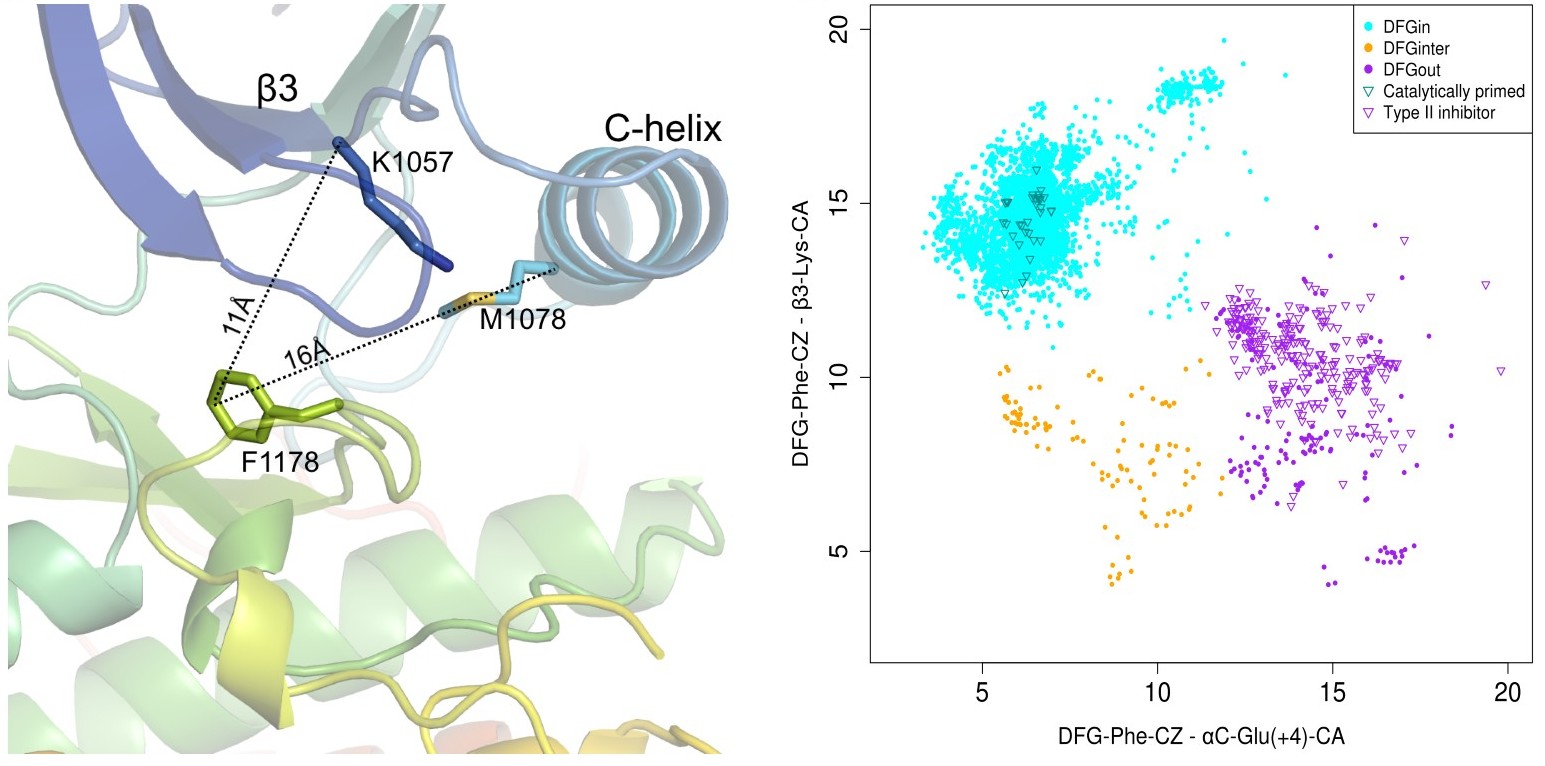
New criteria for active kinase structures (Feb 2026). The new criteria more effectively distinguish substrate-binding
capability at the C-terminal segment of the activation loop, compared to the 2023 criteria (described in
the 2023 biorxiv preprint). With these new criteria, we have produced AlphaFold2 active structures of
all 437 catalytically active human kinases (i.e., excluding pseudokinases)
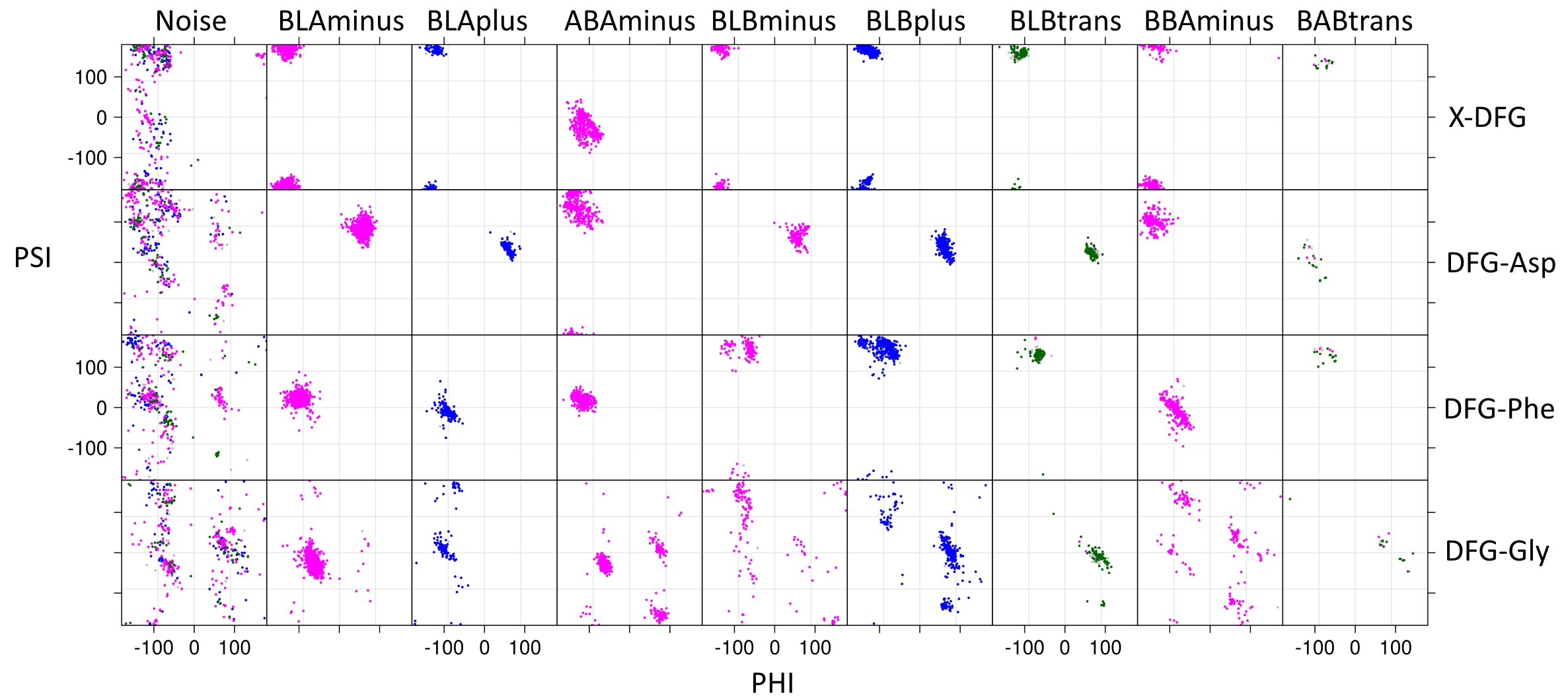
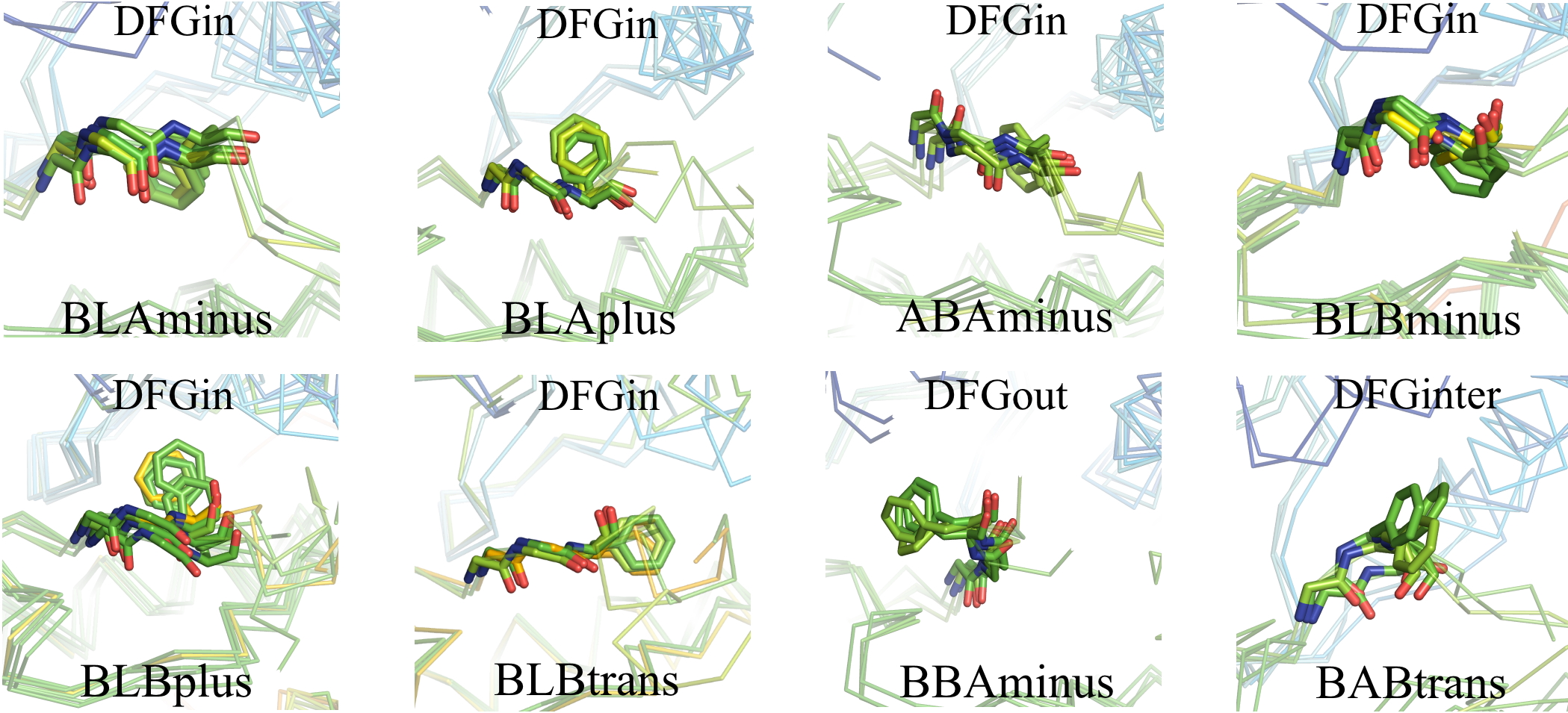
All structures have an ‘Activity’ label based on six criteria: DFGin, BLAminus, the N-terminal domain salt bridge, the HRD motif conformation, and the positions of the N and C terminal segments of the activation loop. Labels corresponding to these criteria are listed in the columns headed by ‘Spatial label,’ ‘Dihedral label,’ ‘Chelix-Saltbridge label,’, ‘HRD label’ and ‘ActLoop label’ presented on every page. Structures must be DFGin, BLAminus, HRD-in, and Saltbr-in to be considered "Active". If atoms are missing, then the structure is considered Inactive. In addition, the ActLoop-NT and ActLoop-CT labels are used to determine if the structure can bind substrates. If residues are missing, then the kinase activity label is "None". If any of these criteria are "out", then the structure is labeled Inactive. Details will be presented in the updated 2026 preprint (above)

The Chelix-Saltbridge label is composed of two components: (1) The Chelix is labeled ‘in’ or ‘out’ (or ‘none’ if residues are missing), based on the distance of the Cbeta atoms of the Lys and Glu of the N-terminal domain (≤10 Å means ‘in’, >10 Å means ‘out’). (2) The saltbridge is ‘in’ if the shorter of the NZ-OE1 and NZ-OE2 distances is ≤3.6 Å, and otherwise is ‘out’. (‘none’ if atoms are missing).
The HRD label is ‘in’ is based on the backbone dihedral angles of the His and Arg residues, which must be in the alpha and L regions of the Ramachandran map, respectively.
The ActLoopNT label is ‘in’ if the there is a hydrogen bond (≤3.6 Å) between the N or O atoms of the sixth residue of the activation loop (DFGxxX) and the O or N atoms of the residue before the HRD motif (Xhrd). Otherwise it is ‘out’ (or ‘none’ if the atoms are missing). This criterion ensures that the N-terminal segment of the activation loop is extended along the surface of the kinase so that substrate can bind properly.
The ActLoopCT label is ‘in’ if the structure conforms to a set of dihedral angle constraints and distance constraints on the C-terminal residues of the activation loop. Since TYR kinases bind substrates differently than other kinases, the requirements differ between TYR kinases and nonTYR kinases. The criteria are listed on the "advanced" page for each search result.
For nonTYR kinases, there are dihedral angle requirements on the backbone of residues APE6, APE7, and APE8 (counting backwards from the end of the activation loop: e.g., APE6 is XxxAPE). APE7-APE6 have to be in the A,A regions of the Ramachandran map or in the B,L regions, where:
B=φ ∈ (-180°, 0°), ψ ∈ (50°, 180°)
L=φ ∈ (0°, 180°), ψ ∈ (-50°, 100°)
APE8 has to be in the B region of the Ramachandran map. The side-chain rotamer of APE8, when this residue is Ser or Thr, should be gauche-minus (χ1 ∈ (-120°, 0°)). The rotamer requirement is utilized to confirm a hydrogen bond between the side-chain of APE8 and the side-chain of the HRD aspartic acid. There are further distance requirements:
APE10(Cβ)-DFG4(Cα)-dis ∈ (7 Å, 14 Å)
APE11(Cβ)-DFG4(Cα)-dis ∈ (8 Å, 14 Å)
APE12(Cβ)-DFG4(Cα)-dis (<8 Å)
For TYR kinases, there are dihedral angle requirements on the backbone of residues APE6, APE7, APE8, APE9, and APE10. The requirements on APE6-APE7 ensure an alpha-helical formation, while those on APE8-APE10 ensure a beta-strand formation with substrates. There is also a distance constraint between APE9 Cα and the carbonyl oxygen of the HRD residue.
The ActLoop label is a composite of these two labels.
In the Pymol sessions and coordinates, the Activity label follows the kinase+species name. The Saltbridge and ActLoop NT+CT criteria are contained in a string consisting of ‘SNC’ followed by one-letter states (‘i’ for ‘in’; ‘o’ for ‘out’; ‘n’ for ‘none’). For example, ‘SNCiio’ means the Saltbridge is ‘in’, the ActLoopNT segment is ‘in’;, and the ActLoopCT segment is ‘out’;. This is followed by the APEdihe and APEdist labels. APEdihe covers the dihedral angles of APE10-APE9-APE8-APE8rot-APE6+7. APEdist consists of the distance labels for residues APE12-APE11-APE10-APE9.
We have added AlphaFold2 models of active forms (given the criteria above) for all
437 catalytically competent human kinases with typical kinase domains (i.e., excluding 56 pseudokinases).
These are all accessible through the search page for family, gene, etc. Each AlphaFold2 model
is labeled with the form ‘AF-P12345-K1’, where ‘P12345’ is the Uniprot code,
and ‘K1’ means the first Kincore model we have included in the database for that kinase.
See this page to download all the models and associated data.