Conformation-dependent library for backbone geometry in proteins
Description
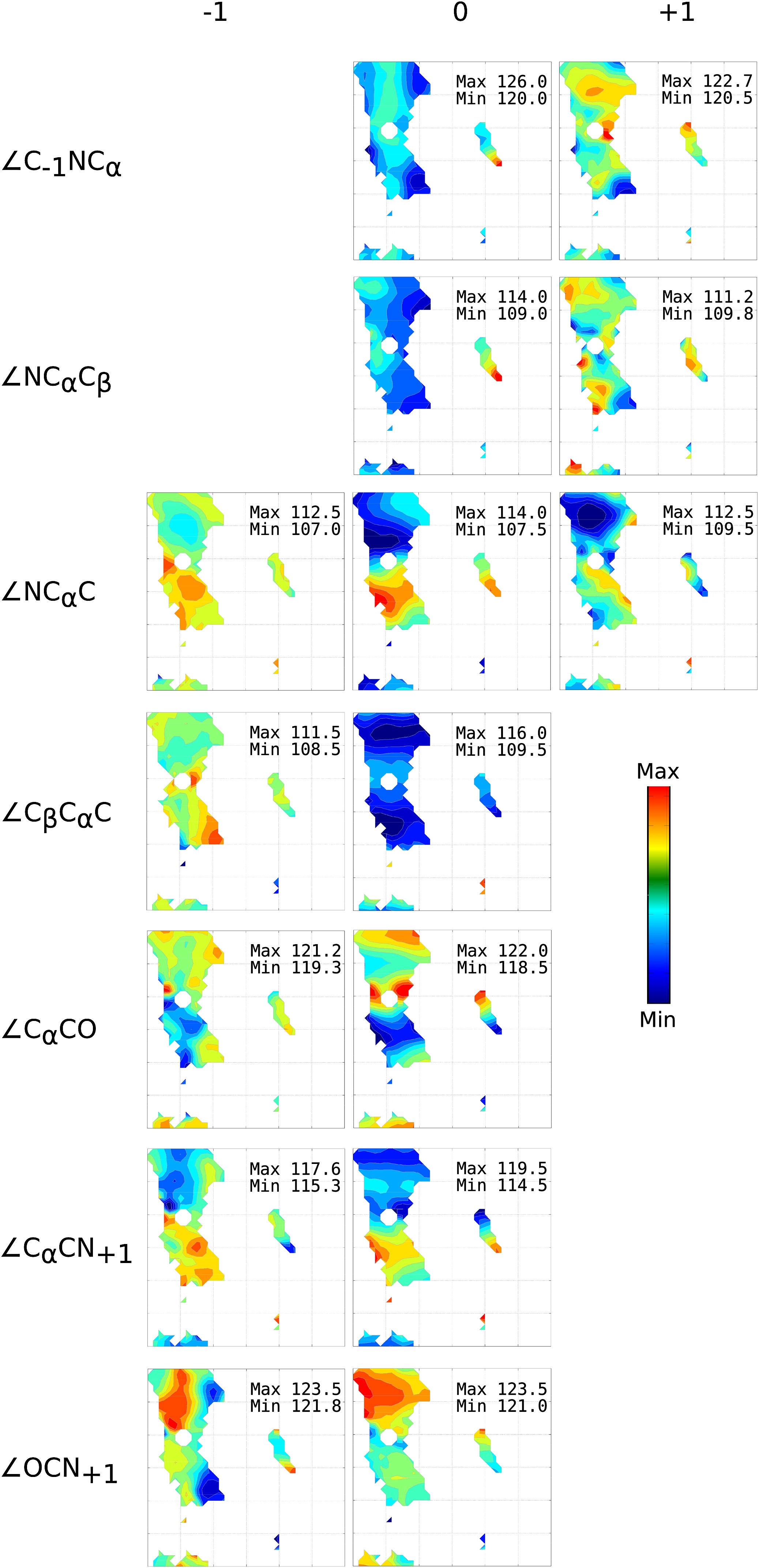
This library contains information on the variation of backbone bond angles and bond lengths as a function of the backbone dihedral angles φ and ψ. For some parameters, there is a systematic variation with φ or ψ or both. This study was initiated by P. Andrew Karplus at Oregon State University. We performed kernel regression analysis to produce smoothly varying regression values for bond lengths and bond angles on φ and ψ.
You can download the conformation-dependent backbone geometry library in an uncompressed, text format or in a zip archive. The text format is both human- and computer-friendly. The library file has Quick-Start information in its header.
The publication of this work (PDF with supplemental figures) can also be found at Structure (2009), and its supplemental figures here.
For more information, visit our collaborators' websites:
proteingeometry.sourceforge.net
pgd.science.oregonstate.edu
Caption: A Ramachandran plot is shown for each backbone bond angle in the two peptide units surrounding the central residue of the tripeptide. The seven unique peptide bond angles are associated with either residue −1, 0, or +1 based on which residue contributes at least two atoms to the angle. ϕ and Ψ in each plot, however, refer to the central residue, 0. Within each plot, colors indicate averages ranging from the global minimum (blue) to the global maximum (red) as calculated using kernel regressions. The global minima and maxima are provided in each plot.
Download FigureAbstract
Protein structure determination and predictive modeling have long been guided by the paradigm that the peptide backbone has a single, context-independent ideal geometry. Both quantum-mechanics calculations and empirical analyses have shown this is an incorrect simplification in that backbone covalent geometry actually varies systematically as a function of the φ and ψ backbone dihedral angles. Here, we use a nonredundant set of ultrahigh-resolution protein structures to define these conformation-dependent variations. The trends have a rational, structural basis that can be explained by avoidance of atomic clashes or optimization of favorable electrostatic interactions. To facilitate adoption of this paradigm, we have created a conformation-dependent library of covalent bond lengths and bond angles and shown that it has improved accuracy over existing methods without any additional variables to optimize. Protein structures derived from crystallographic refinement and predictive modeling both stand to benefit from incorporation of the paradigm.
Acknowledgments
This work was funded under grant P20 GM076222 from the National Institutes of Health and the National Institute for General Medical Sciences.