Servers
ProtCID – a database of clustered protein interactions
ProtCID is a database of similar protein-protein interfaces in crystal structures of homologous proteins. Its main goal is to identify and cluster homodimeric and heterodimeric interfaces observed in multiple crystal forms of homologous proteins. Such interfaces, especially of non- identical proteins or protein complexes, have been associated with biologically relevant interactions. We cluster protein interfaces at both the full-chain level and the domain level, so that both intra- and interprotein interactions are captured. ProtCID also provides clusters of domain-peptide, domain-ligand, and domain-nucleic-acid structures.
PISCES – a protein sequence culling server

PISCES provides non-redundant subsets of protein chains in the PDB according to user-input criteria such as resolution, R-factor, chain length, and pairwise sequence identity between chains. We calculate the sequence identity between all homologous proteins in the PDB with alignment of hidden Markov models of each unique sequence. PISCES also allows the user to input their own list of PDB entries or PDB chains, so that other user-defined criteria can be imposed before the pairwise sequence identity, which results in the longest possible lists.
PDBrenum – Renumbering of protein structures according to UniProt sequences
The amino acids in protein structures in the PDB can be numbered in any way that authors may choose, and different structures of the same protein may be numbered differently. We present a server and program for renumbering PDB structures according to the UniProt sequence for each chain in the structure. The server provides searches by PDB entry and UniProt code (P38398 or BRCA1_HUMAN) and allows the user to download renumbered structures of the asymmetric unit and biological assemblies in both mmCIF and legacy-PDB format.
PyIgClassify2
PyIgClassify is a web server primarily providing the clusters and associated information of the antibody complementarity determining regions (CDRs) in the Protein Data Bank (PDB). Our clustering is based on a dihedral-angle metric between pairs of CDR structures of a given length. The associated nomenclature is a simple combination of the CDR name, the length, and the cluster number (ranked from largest to smallest), e.g. L1-11-1. This clustering is used in our design program, RosettaAntibodyDesign.
Kincore
Kincore presents a classification of the structures of active and inactive protein kinases from our clustering of kinase conformations published in PNAS. The clustering is based on the location of the Phe side chain (DFGin, DFGout, and DFGinter or intermediate) and the backbone dihedral angles of the sequence X-D-F, where X is the residue before the DFGmotif, and the DFG-Phe side-chain rotamer, utilizing a density-based clustering algorithm. We have identified eight distinct conformations and labeled them based on the Ramachandran regions (A, alpha; B, beta; L, left) of the XDF motif and the Phe rotamer (minus, plus, trans). Our clustering divides the DFGin group into six clusters including BLAminus, which contains active structures, and two common inactive forms, BLBplus and ABAminus. DFGout structures are predominantly in the BBAminus conformation, which is essentially required for binding type II inhibitors. The server provides conformational labels and ligand types (Type I, II, etc.) for every kinase structure in the PDB and can be searched by gene, kinase family, PDB entry, and ligand.
Software
Betaturns
BetaTurnLib18 & BetaTurnTool18 Library of 18 β turns and their Assignment Tool for mmCIF and PDB input. Supported on Unix / Mac / Windows.
SCWRL4.0

SCWRL4.0 is the most recent version of the SCWRL program for prediction of protein side-chain conformations. SCWRL4.0 is based on an algorithm based on graph theory that solves the combinatorial problem in side-chain prediction more rapidly than many other available program.
SecNet – Protein Secondary Structure Prediction
We developed a program with convolutional neural networks for predicting the secondary structure of proteins from sequences. With rigorous benchmarking on a test set with no related proteins in the training or validations sets, our program achieves an 84% accuracy for three- label predictions (helix, sheet, coil) and a 74% accuracy on eight-label predictions (H,E,C,T,B,S,I,G).
Libraries
Backbone-dependent rotamer library
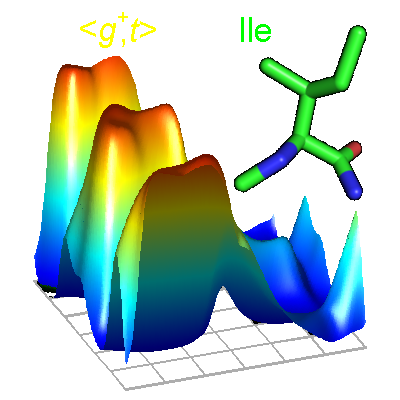
The backbone-dependent rotamer library expresses the probability and mean dihedral angles of each rotamer of each protein residue type as a function of the backbone dihedrals phi and psi. It has been widely used in protein structure prediction and protein design programs such as Rosetta. The latest library is based on over 4000 high-resolution structures and was determined using adaptive kernel density estimates and kernel regressions.
Neighbor-dependent Ramachandran maps
We provide Ramachandran probability distributions for residues in protein loops from a high- resolution data set with filtering based on calculated electron densities. Distributions for all 20 amino acids (with cis and trans proline treated separately) have been determined, as well as 420 left-neighbor and 420 right-neighbor dependent distributions.
Omega dihedral angle regressions
The planarity of peptide bonds is an assumption that underlies decades of theoretical modeling of proteins. However, empirical analyses of atomic-resolution protein structures reveal that trans peptide groups can vary by more than 25° from planarity. Analyses as a function of the φ,ψ- backbone dihedral angles show that the expected value deviates by ± 8° from planar as a systematic function of conformation. To account for the systematic φ,ψ-dependent component of nonplanarity, we present a conformation-dependent library that can be used in crystallographic refinement and predictive protein modeling.
Protein backbone bond angle regressions
Protein structure determination and predictive modeling have long been guided by the paradigm that the peptide backbone has a single, context-independent ideal geometry. Empirical analyses have shown this is an incorrect simplification in that backbone covalent geometry actually varies systematically as a function of the phi and psi backbone dihedral angles. We used a nonredundant set of ultrahigh-resolution protein structures to define these conformation- dependent variations and to perform regressions of thee angles against the backbone dihedrals phi and psi. The trends have a rational, structural basis that can be explained by avoidance of atomic clashes or optimization of favorable electrostatic interactions. We have created a conformation-dependent library of covalent bond lengths and bond angles.
Seq2Coord (S2C)
Because authors can number amino acids in a structure in different ways, it is useful to map the sequence of each chain with amino acids numbered from 1 to N to the residues in the coordinates. S2C are simple files that provide this mapping and are meant to be used in conjunction with legacy-PDB format files. mmCIF files provide this information automatically.