Smooth Backbone-Dependent Rotamer Library 2010
Tutorial for 2010 smooth backbone-dependent rotamer library
IntroductionBasicsTorsion or Dihedral Angle For A Group of Four AtomsA torsion angle or dihedral angle is the angle between two planes. For four consecutively bonded atoms A-B-C-D, atoms A-B-C define the first plane and atoms B-C-D define the second plane. The angle between these two planes (A-B-C and B-C-D) is a dihedral or torsion angle. The positive rotation is the clockwise rotation of the vector C-D relative to the vector A-B when looking in the direction of the B-C vector: 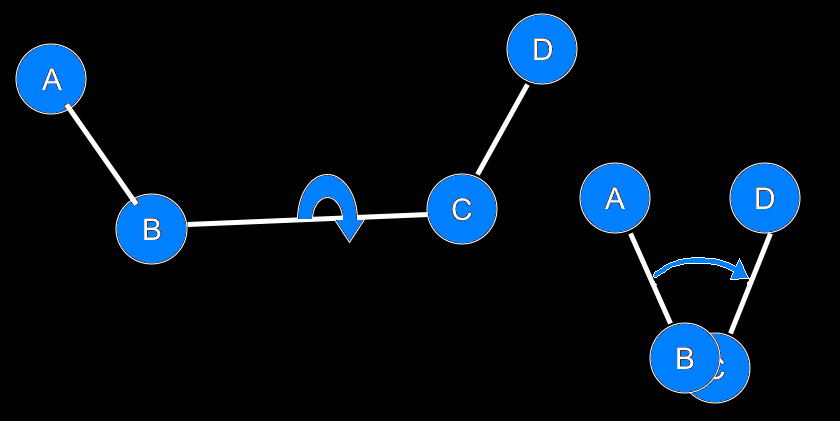 When all 4 atoms lie in the same plane and A and D atoms are on the same side relative to B-C vector, the torsion angle is zero: 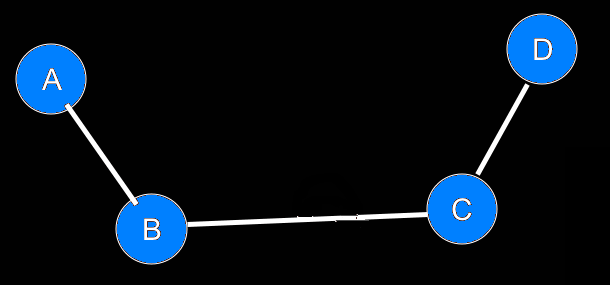 If they are on opposite sides then the angle is 180° or -180°: 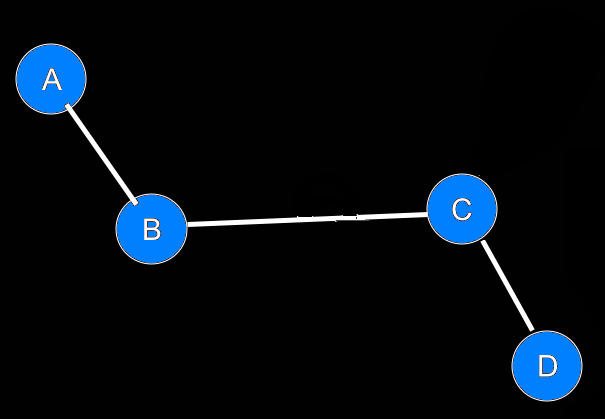 The torsion angle, A-B-C-D only alters the distance between atoms A and D; the other interatomic distances are constrained by approximately constant bond lengths and bond angles. Protein Backbone Torsion Angles, φ and ψThe backbone torsion angles are ω, φ and ψ. The φ dihedral angle for residue i is defined by Ci-1-Ni-Cαi-Ci; the ψ dihedral angle for residue i is defined by Ni-Cαi-Ci-Ni+1; the ω dihedral angle for residue i is defined by Cαi-1-Ci-1-Ni-Cαi. ω is almost always near 180°; although there is some variation dependent on the values of ψi-1 and φ. For more details, click here. 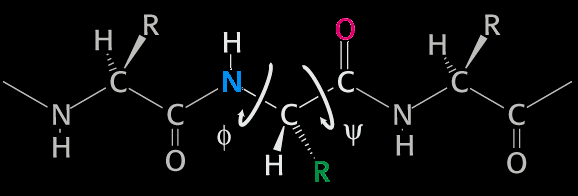 Protein Side-Chain Torsion Angles, χ1, χ2, χ3 and χ4The side-chain torsion angles, χ1, χ2, χ3 and χ4 define a side-chain conformation. For example, in the case of lysine, χ1 is N-Cα-Cβ-Cγ and defines a rotation around the Cα-Cβ bond. χ2 is Cα-Cβ-Cγ-Cδ and defines a rotation around the Cβ-Cγ bond. χ3 is Cβ-Cγ-Cδ-Cε and defines a rotation around the Cγ-Cδ bond. χ4 is Cγ-Cδ-Cε-Nζ and defines rotation around the Cδ-Cε. 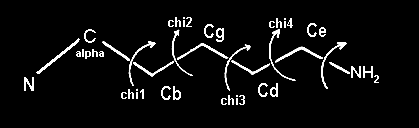 Traditional Rotamer LibraryRotamers, rotameric χMost side-chain χ torsion angles, are centered on sp3-sp3 hybridized bonds, and exhibit three narrow, approximately symmetric peaks in their probability density distributions. For example, here is the backbone-independent density for methionine χ1: 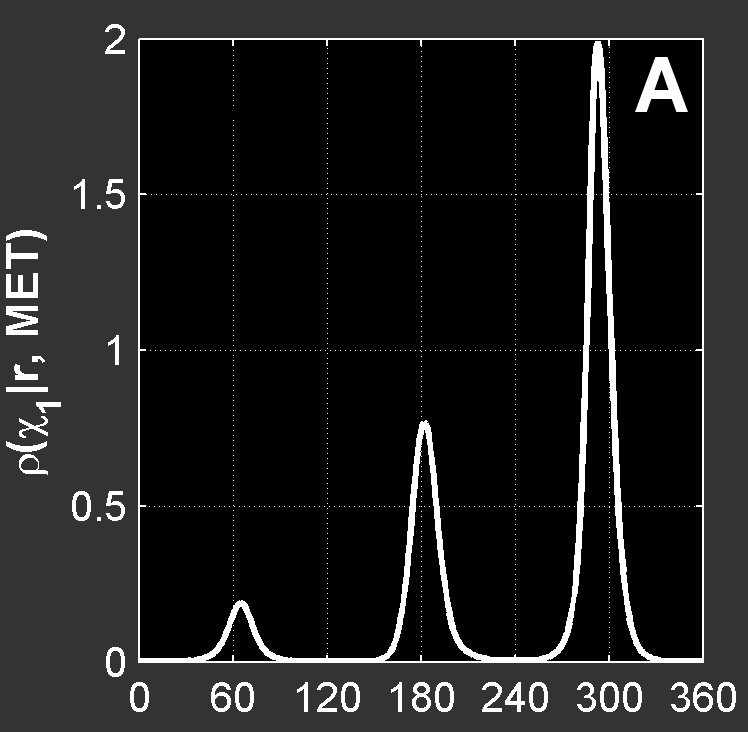 MET χ1 has gauche+ (g+), trans (t), and gauche- (g-) peaks at approximately 60°;, 180°, and 300° respectively. 300° for g- is the same as -60°. The location and shape of these peaks vary somewhat (by at most 20°) a residue backbone conformation changes, and their standard deviations are at usually less than 10° in high-resolution structures. It therefore makes sense to define a set of discrete side-chain conformations (χ1, χ2, χ3 and χ4), also known as rotamers. We refer to such χ degrees of freedom as rotameric. The prevailing majority of side-chain χ angles are rotameric, comprising the following: ARG χ1, χ2, χ3, χ4 HIS χ1 PRO χ1
ASN χ1 ILE χ1 SER χ1
ASP χ1 LEU χ1, χ2 THR χ1
CYS χ1 LYS χ1, χ2, χ3, χ4 TRP χ1
GLN χ1, χ2 MET χ1, χ2, χ3 TYR χ1
GLU χ1, χ2 PHE χ1 VAL χ1
Non-rotameric χ.Nevertheless, not all side-chain χ angles adhere to a concept of a rotamer. Non-rotameric degrees of freedom in protein side chains are centered on sp3-sp2 bonds, and exhibit broad and often asymmetric probability density distributions. These non-rotameric degrees of freedom exist in ASN, ASP, GLN, GLU and the aromatic amino acids PHE, TYR, HIS and TRP. Each of these residue types has one or two (in the case of GLN and GLU) rotameric degrees of freedom nearer the backbone, while the terminal degree of freedom is non-rotameric. The shape and location of the distributions of these non-rotameric dihedral angles vary depending on the backbone conformation and the rotamer of the non-terminal degrees of freedom. As examples, we show the backbone-independent χ2 probability densities for asparagine and tryptophan for each of the three χ1 rotamers (<g+>, <t>, <g->) of these residue types and χ3 densities for all 9 χ1, χ2 rotamers of glutamine (<g+, g+>, <g+, t>, ... and <g-, g->): 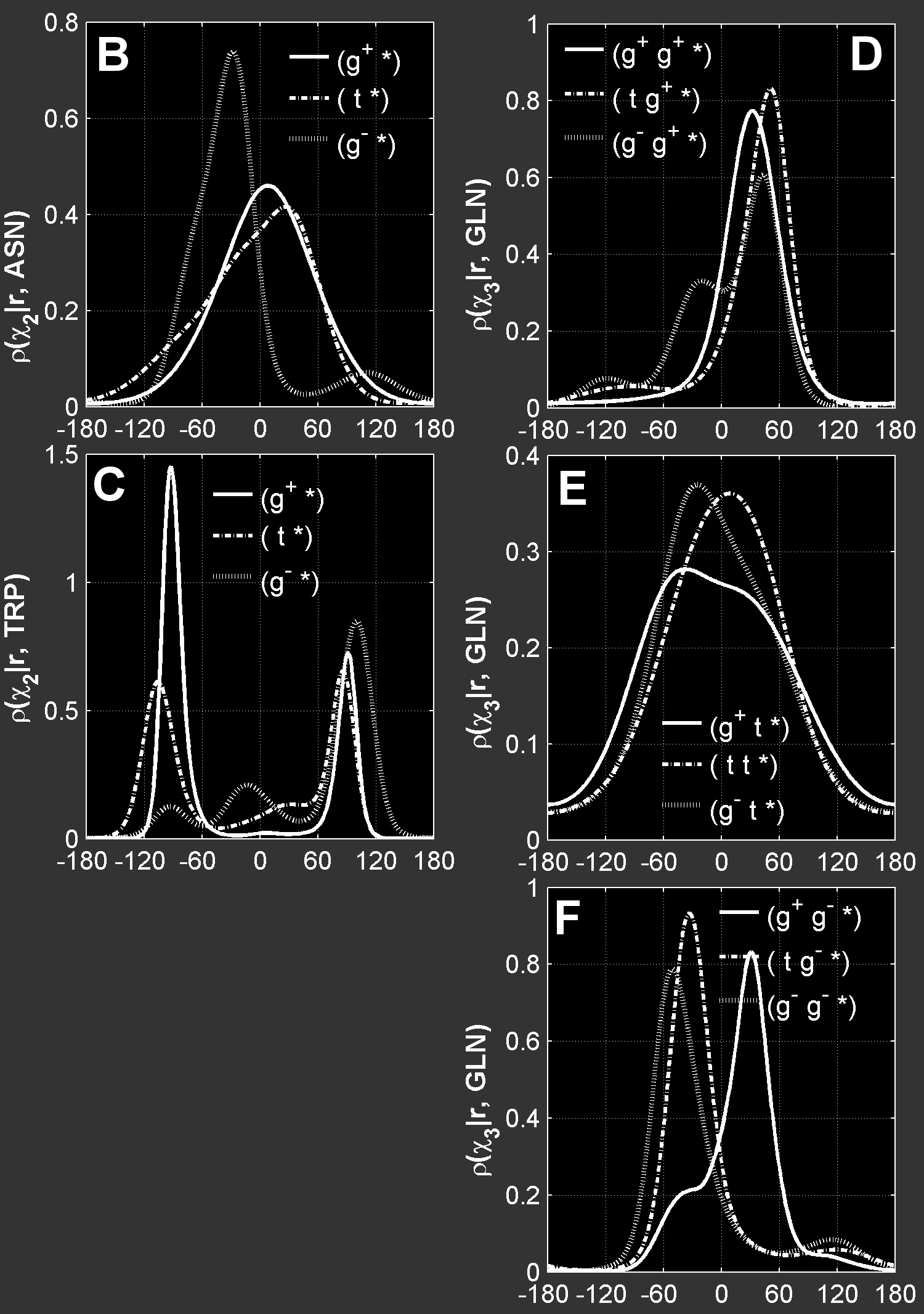 Here is a gif animation illustrating how χ2 density varies as a function of φ and ψ for the trans χ1 rotamer of ASN. The Ramachandran density for the trans χ1 of ASN is shown in the inset in the top-right corner, ρ(φ, ψ | r = <t>) showing the major secondary-structure conformational regions: α-helix, antiparallel β-sheet, parallel β-sheet and left-handed helix. A brighter color corresponds to a higher density. The red box on the inset figure and the numeric values in the title indicate the current backbone conformation (φ, ψ). As the backbone conformation changes we can see several different χ2 populations dominating in different (φ, ψ) regions. The positions of their maximum values move with φ and ψ and sometimes they coalesce into a single wide population. You can watch movies for all non-rotameric degrees of freedom here. ASN, r = <t>The non-rotameric degrees of freedom are: ASN χ2 GLN χ3 PHE χ2 HIS χ2
ASP χ2 GLU χ3 TYR χ2 TRP χ2
Traditional backbone-dependent rotamer libraryUnder the term "traditional rotamer library", we mean a library of discrete side-chain conformations only, i.e. rotamers. The traditional rotamer library includes: rotamer frequencies, their mean torsion angles and standard deviations. These values vary significantly as a residue backbone conformation changes. Therefore, a traditional backbone-dependent rotamer library contains values for rotamer frequencies, mean χ angles and standard deviations as a function of the backbone torsion angles, φ and ψ. In this traditional rotamer model, each side-chain χ has a set of discrete conformations, i.e. rotamers. For instance, serine has only one degree of freedom, χ1. Its χ1 has 3 rotamers with mean values about +60°, 180° and -60°. Therefore, for serine there are 3 rotamers in total: <g+>, <t> and <g->. In contrast, leucine has two side-chain degrees of freedom, i.e. χ1 and χ2. Each of them has its own 3 rotamers, g+, t and g-, producing 3 × 3 = 9 rotamers in total: <g+, g+>, <g+, t>, <g+, g->, <t, g+>, <t, t>, <t, g->, <g-, g+>, <g-, t> and <g-, g->. We can designate g+, t and g- rotamers as 1, 2 and 3 respectively for serine χ1, leucine χ1 and leucine χ2. Thus serine has <1>, <2> and <3> rotamers while leucine has <1, 1>, <1, 2>, ..., <3, 3>. We can use the same number designations for the rotamers of the 18 standard amino acids with flexible side chains. For example, arginine and lysine have 3 × 3 × 3 × 3 = 81 rotamers in total (<1, 1, 1, 1>, <1, 1, 1, 2> ... <3, 3, 3, 3>). For the non-rotameric degrees of freedom, the angular space can be divided into some number of bins to approximate a rotamer model. For example, for ASN χ2 in 1997 we used three rotamers to represent the dihedral angle distribution. In 2002, we expanded this to six rotamers for χ2, so ASN had 3 × 6 = 18 (<1, 1>, <1, 2>, ... <3, 6>) rotamers. For the 2010 rotamer names, definitions and total counts of 18 amino acids please refer to the table below: Rotamer library data and Rotamer definitions }}) * TPR (trans proline), CPR (cis proline) and CYH (non-disulfide-bonded Cys), CYD (disulfide-bonded Cys) percentages are calculated relative to the total number of PRO and CYS respectively. Each rotameric degree of freedom, χ has the rotamer definitions: g+ = [0°, 120°), t = [120°,240°), g- = [240°,360°). † PRO, CPR, and TPR have only g+ and g- rotamers. Rotameric Vs Non-rotameric χ anglesIn the new, 2010 library, we strictly distinguish between rotameric and non-rotameric degrees of side-chain freedom based on the hybridization state of the atoms involved in a corresponding torsion angle, χ. 2010 LibraryAims and FeaturesThere were several aims in deriving a new backbone-dependent rotamer library:
Methods in a NutshellWe applied adaptive kernel density estimates to compute backbone-dependent probabilities of side-chain rotamers and adaptive kernel regression to estimate backbone-dependent mean χ angles and their standard deviations. The central concept of these methods is to put a bell-shaped curve, i.e. a kernel function on top of each experimental data point from a data set. This action converts a set of discrete data points to a set of continuous objects, i.e. smooth functions. The bell-shaped curve has a bandwidth which locally adapts depending on the amount of available data. Since we are deriving for rotamer probabilities, means and standard deviations as a function of φ and ψ, we used the periodic von Mises probability distribution as the kernel choice rather than Gaussians or other non-periodic kernels. For the non-rotameric degrees of freedom, we developed an innovative adaptive kernel regression of adaptive kernel density. Here we place a kernel on top of each experimental data point not only in φ and ψ space but also in χ space in such a way that for any φ and ψ a combination of χ kernels leads to a local estimate of χ probability density. We chose an appropriate method to adapt these kernels in the φ,ψ space and χ space as well to produce statistically sound estimates and at the same time not to lose local details of the side chain model. For a complete description of the methods, please click here. Continuous functions Vs Subsequent Discretization on a GridWe developed methods that enable estimation estimation of rotamer statistics or non-rotameric χ density as continuous function of φ and ψ, i.e. at any numerical values of φ and ψ. However, we evaluated the rotamer probabilities and angle statistics on a 10° grid in φ and ψ in order to provide a rotamer library that would be compatible with methods that require a fast lookup of these values, such as Rosetta or SCWRL. The text files (which can be converted to binary files) are a good trade-off between memory overhead and experimental precision in structures. The advantages are backward compatibility, relatively low memory overhead, no CPU overhead, no restriction to a particular platform, operating system, computer language. There are several disadvantages. Users have to implement their own parsers. There is reduced accuracy of the estimated statistics at points not on the grid and decreased accuracy of derivatives when these are calculated from neighboring grid values. In the future, we will provide code that produces rotamer statistics at any value of φ and ψ, but this requires significant optimization and accurate numerical methods for derivative computation, etc. 2010: Traditional Rotamer Model Vs. New Model of χ DensitiesIn 2010 we provide two different types of backbone-dependent side-chain models: 1) the traditional rotamer model as in 1997 and 2002 for both rotameric and non-rotameric degrees of freedom (in 30° bins); 2) the traditional model for the rotameric degrees of freedom but also full probability density estimates for the non-rotameric degrees of freedom as a function of φ and ψ. The traditional model provides support for existing applications. Its format has remained the same as the 2002 libraries. While the traditional 2010 library demonstrates improved performance over the 2002 library, there are applications where a backbone-dependent model of χ densities may provide increased accuracy. It is also possible to use both, the traditional rotamer library in the first stage of modeling and full χ densities at the second, refinement stage. Download Options for 2010 "Traditional Rotamer Library" PackagesFor a definition and description of Traditional Rotamer Library, see above. The traditional backbone-dependent rotamer library is available as a single larger file. There are 18 standard amino acid types with (glycine and alanine do not have flexible side chains). There are three choices to make:
A user decides whether additional subclasses are needed for proline and cysteine in their specific applications. While the differences between trans PRO and cis PRO or between non-disulfide-bonded and disulfide-bonded CYS are not as drastic as between different residue types, they are still significant and may lead to improved accuracy. As discussed above, the density distributions for the non-rotameric χ angles demonstrate backbone dependence, i.e. the shape and location of the density peaks significantly vary as φ and ψ change. For this reason the "rotamer" definitions, i.e. their left and right limits vary as a function of φ and ψ. We provide two options. The first option has dynamic or backbone-dependent "rotamer" definitions for the 8 non-rotameric χ angles. The second option has static or backbone-independent "rotamer" definitions. In applications where a user does not care about assigning a rotamer type to an experimental side-chain conformation or does not need to calculate rotamer-specific derivatives, the first option with backbone-dependent "rotamer" definitions is preferable, since the mode of the distribution is centered in the maximum probability bin. In contrast, for the applications where a user wants to query a likelihood of some side-chain conformation or wants a rotamer, for example GLN <1,3,7> to remain the same, i.e. with the same left and right limits, the second choice with the backbone-independent definitions is desirable. We provide 8 small files for each non-rotameric χ with these backbone-independent definitions. As shown above, the statistical methods used in the 2010 library are completely different from the ones used in 1997 or 2002. The 1997 and 2002 libraries relied on Bayesian formalism while the 2010 library takes advantage of adaptive kernel density estimates, kernel regressions and adaptive kernel regressions of densities. These kernel methods were chosen to produce smoother and more accurate libraries than in 1997/2002. The level of smoothness was separately optimized for each residue type, each degree of freedom and each library component. However, the library consists of many different components and the final performance is an interplay of separate components. Our benchmark tests in SCWRL4 and Rosetta demonstrated the additional 5% smoothness lead to improved accuracy in these applications. The 5% library is our suggested, default choice. Nevertheless, there are some applications where a user can benefit from increased or decreased smoothness. Additional testing is required for such applications. For example, there is a scenario when a user may introduce the smoothest, 25% library in initial modeling and at the final stage of modeling during the refinement switch to 0% library with the greatest amount of details. New Model of χ DensitiesFor the 8 non-rotameric χ angles, we developed a new model describing their internal properties in a statistically more appropriate way. Each non-rotameric χ is modeled with one-dimensional probability density distribution varying as a function of the backbone φ and ψ. The non-rotameric χ is always the last χ in these side-chain types, i.e. χn; the preceding χ angles are rotameric, i.e. χ1, χ2, ... χn-1. There are therefore Ntot = N1 × N2 × ... × Nn-1 rotamers for each side chain with a non-rotameric degree of freedom. Separately for each of the Ntot rotamers, we estimated the χn probability density as a function of φ and ψ, i.e. ρ(χ-n | r, φ, ψ). The remaining residue types with all rotameric χ angles are modeled as a Traditional Rotamer Library. Their rotamer model allows for quick traversal through discrete side-chain conformation space which is a good statistical model for sp3-sp3 hybridized bonds. In future we may consider modeling all χ angles (whether rotameric or non-rotameric) for all residue types as backbone-dependent χ densities along with the traditional rotamer model which obviously has advantages in exhaustive searches of conformation space. Download Options for 2010 "Model of χ Densities" PackagesThe new model of χ densities has separate file(s) for each residue type. For each rotameric residue type there is always a single file with backbone-dependent rotamer probabilities (Traditional Rotamer Model). For each non-rotameric residue type we provide three files. The first file contains the χ Densities. The second and third files purely belong to Traditional Rotamer Model. They are provided for convenience, so that the user can easily switch between two models for the non-rotameric residue types if needed.
A user can choose additional smoothness applied to all components of a rotamer library at the level of
FormatGeneralDepending on what library model a user chooses from a list, several files in different text formats are available from a distributive package. The names and formats of the files are self-explanatory. A header of each file describes what is included in a file and some important options and their values used in a generated library. When parsing a file, a user can always skip any commentary information by ignoring lines starting with "#": # Backbone-dependent rotamer library with regular rotamers
#
# phi interval, deg [-180.0, 180.0]
# phi step, deg 10.0
# psi interval, deg [-180.0, 180.0]
# psi step, deg 10.0
#
# Residue type MET
#
# Rotamer probability precision 0.000001
#
# Number of chi angles (degrees of freedom) 3
# Number of chi angles treated as discrete 3
# Number of bins for each discrete chi angle [3, 3, 3]
# Number of rotamers for discrete chi angles 27
# Number of chi angles treated as continuous 0
#
# TotalDatapointsNum 12240
The actual data is included on the lines not starting with "#". The library data are presented in a table form either space or tab delimited. The beginning of the table is preceded with a "#", commentary line showing self-descriptive titles for each of its columns: ser.bbdep.rotamers.lib
# T Phi Psi Count r1 r2 r3 r4 Probabil chi1Val chi2Val chi3Val chi4Val chi1Sig chi2Sig chi3Sig chi4Sig
#
SER -180 -180 19 1 0 0 0 0.802596 68.0 0.0 0.0 0.0 8.1 0.0 0.0 0.0
SER -180 -180 19 2 0 0 0 0.197211 -175.4 0.0 0.0 0.0 10.1 0.0 0.0 0.0
SER -180 -180 19 3 0 0 0 0.000193 -62.9 0.0 0.0 0.0 8.6 0.0 0.0 0.0
asn.bbind.chi2.Definitions.lib
# r1 r2 r3 r4 P -logP left chi2 right
1 1 0 0 0.236 1.443 -7.000 7.526 23.000
1 2 0 0 0.195 1.634 23.000 36.529 53.000
asp.bbdep.densities.lib
# T Phi Psi Count r1 Probabil chi1Val chi1Sig -90 -85 -80 ... 85
#
ASP -80 -10 404 3 0.630735 -68.8 7.7 4.505e-003 6.261e-003 8.664e-003 ... 3.256e-003
ASP -80 -10 404 1 0.342077 62.7 7.2 8.006e-003 1.022e-002 1.289e-002 ... 6.260e-003
ASP -80 -10 404 2 0.027188 -166.9 11.9 2.047e-002 1.350e-002 8.825e-003 ... 3.024e-002
"Traditional Rotamer Model" PackageMain File Formatwith rotamer probabilities, mean and sigma of rotameric χ anglesWhen the 2010 library is used according to a traditional model of discrete conformations, i.e. rotamer model, there is a large text file with merged data for either 18 or 22 residue types (see above). We preserved the original format of the 1997 or 2002 text libraries in order to support older, existing applications: ALL.bbdep.rotamers.lib
# T Phi Psi Count r1 r2 r3 r4 Probabil chi1Val chi2Val chi3Val chi4Val chi1Sig chi2Sig chi3Sig chi4Sig
#
LEU -70 -40 5884 3 2 0 0 0.668530 -68.3 173.0 0.0 0.0 6.5 8.1 0.0 0.0
LEU -70 -40 5884 2 1 0 0 0.296974 -177.4 58.8 0.0 0.0 7.2 6.1 0.0 0.0
LEU -70 -40 5884 3 1 0 0 0.014762 -88.5 60.0 0.0 0.0 7.7 10.2 0.0 0.0
LEU -70 -40 5884 2 2 0 0 0.009079 -175.1 153.0 0.0 0.0 9.3 10.0 0.0 0.0
LEU -70 -40 5884 3 3 0 0 0.007372 -89.0 -62.2 0.0 0.0 9.2 16.8 0.0 0.0
LEU -70 -40 5884 2 3 0 0 0.001641 -174.6 -78.6 0.0 0.0 9.5 11.9 0.0 0.0
LEU -70 -40 5884 1 1 0 0 0.001614 72.2 85.1 0.0 0.0 6.8 7.0 0.0 0.0
LEU -70 -40 5884 1 3 0 0 0.000020 70.3 -63.0 0.0 0.0 9.2 21.3 0.0 0.0
LEU -70 -40 5884 1 2 0 0 0.000009 72.1 165.9 0.0 0.0 10.1 13.3 0.0 0.0
T - three-letter designation of an amino acid type. Optional File Formatwith backbone-independent "rotamer" definitions for non-rotameric χ anglesFor each of the 8 non-rotameric χn we provide backbone-independent "rotamer" definitions, meaning the definitions are static and do not change as φ and ψ vary. Here is the format description based on glutamic acid: glu.bbind.chi2.Definitions.lib
# r1 r2 r3 r4 P -logP left chi3 right
1 1 1 0 0.406 0.900 5.500 20.276 35.500
1 1 2 0 0.218 1.525 35.500 47.346 65.500
1 1 3 0 0.072 2.631 65.500 78.259 95.500
1 1 4 0 0.041 3.187 -84.500 -70.468 -54.500
1 1 5 0 0.056 2.888 -54.500 -37.366 -24.500
1 1 6 0 0.207 1.575 -24.500 -6.564 5.500
2 1 1 0 0.423 0.860 6.500 21.331 36.500
2 1 2 0 0.237 1.440 36.500 48.692 66.500
2 1 3 0 0.081 2.512 66.500 78.719 96.500
2 1 4 0 0.034 3.377 -83.500 -70.282 -53.500
2 1 5 0 0.038 3.266 -53.500 -36.204 -23.500
2 1 6 0 0.186 1.680 -23.500 -4.851 6.500
3 1 1 0 0.350 1.049 -13.500 1.292 16.500
3 1 2 0 0.240 1.427 16.500 29.564 46.500
3 1 3 0 0.102 2.280 46.500 58.574 76.500
3 1 4 0 0.038 3.273 76.500 89.996 106.500
3 1 5 0 0.063 2.764 -73.500 -55.812 -43.500
3 1 6 0 0.206 1.578 -43.500 -26.206 -13.500
r1 - a numerical designation of a χ1 rotamer, i.e. 1, 2 .. N1. When a residue type doesn't have χ1, 0 is reported. "New Model of χ Densities" PackageRotameric Residue TypesIn the package, "New Model of χ Densities" available for downloading, the rotameric residue types are always modeled with Traditional Rotamer Model. The only difference here is that they are provided as separate files for each type instead of one large merged file. A user can decide on which subclasses of proline or cysteine to use. Non-Rotameric Residue TypesFor each non-rotameric residue types, we provide three files: "χn Densities" File Formatwith densities for non-rotameric χn, rotamer probabilities and mean and sigma of rotameric χ anglestyr.bbdep.densities.lib
# T Phi Psi Count r1 Probabil chi1Val chi1Sig -30 -25 -20 ... 145
#
TYR -60 -40 1769 2 0.558459 -178.1 10.3 6.533e-004 6.394e-004 6.673e-004 ... 7.316e-004
TYR -60 -40 1769 3 0.418968 -72.7 11.3 3.303e-002 3.011e-002 2.714e-002 ... 3.616e-002
TYR -60 -40 1769 1 0.022572 75.1 12.6 6.880e-004 5.504e-004 4.554e-004 ... 9.006e-004
T - three-letter designation of an amino acid type.
A number of columns for r1, r2 ... r(n-1) or chi1Val, chi2Val ... chi(n-1)Val or chi1Sig, chi2Sig ... chi(n-1)Sig is the same as the number of the rotameric χ angles for a residue type. For easier parsing a user can read this number beforehand from the commentary section lines which are in the following fixed format: glu.bbdep.densities.lib
# Number of chi angles (degrees of freedom) 3
# Number of chi angles treated as discrete 2
# Number of bins for each discrete chi angle [3, 3]
# Number of rotamers for discrete chi angles 9
# Number of chi angles treated as continuous 1
The number of the non-rotameric χn integrated probabilities, P[ χn ± ½ × step(χn) ] at the χn values is always 36. The stepsize, starting point and ending point of the χn period interval can be parsed beforehand from either of two places with a fixed format in the commentary section.
glu.bbdep.densities.lib
# chi3 interval, deg [-90.0, 90.0]
# chi3 step, deg 5.0
glu.bbdep.densities.lib
# T Phi Psi Count r1 Probabil chi1Val chi1Sig -90 -85 -80 ... 85
We chose such starting and ending χn because in the 1997 / 2002 libraries some "rotamers" had such staring or ending positions and in addition the χn distributions are better viewed with such limits.
ArticleA smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. |